Overview
Phenylketonuria (PKU) is a rare inherited metabolic disorder in which the body cannot properly break down an amino acid called phenylalanine. Phenylalanine is found in many protein-containing foods and some artificial sweeteners. In people with PKU, the enzyme needed to process phenylalanine is missing or not functioning properly.
As a result, phenylalanine builds up in the blood and brain, which can cause serious health problems if not treated. High levels of this amino acid can damage the developing brain and lead to intellectual disabilities and other neurological issues.
PKU is usually detected through newborn screening shortly after birth. With early diagnosis and careful dietary management, most people with PKU can lead healthy lives and avoid severe complications.
Symptoms
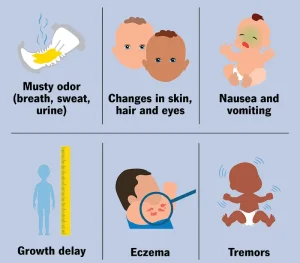
Symptoms of phenylketonuria may not be noticeable in newborns at first but can develop as phenylalanine levels increase in the body.
Common symptoms may include:
-
Developmental delays
-
Intellectual disability if untreated
-
Behavioral problems
-
Hyperactivity
-
Seizures
-
Skin rashes such as eczema
-
Musty or mold-like body odor
-
Lighter skin, hair, and eye color compared to family members
-
Poor growth
Early treatment can prevent many of these symptoms from developing.
Causes
Phenylketonuria is caused by a genetic mutation that affects the gene responsible for producing an enzyme called phenylalanine hydroxylase. This enzyme normally converts phenylalanine into another amino acid called tyrosine.
When the enzyme does not work properly, phenylalanine accumulates in the body. The condition is inherited in an autosomal recessive pattern, meaning a child must inherit the altered gene from both parents to develop PKU.
Parents who carry one copy of the gene usually do not show symptoms but can pass the gene to their children.
Risk Factors
The primary risk factor for phenylketonuria is genetic inheritance.
Risk factors include:
-
Having parents who both carry the PKU gene mutation
-
Family history of phenylketonuria
-
Certain ethnic backgrounds where the condition may be more common
Because PKU is inherited, it cannot be acquired later in life through lifestyle or environmental factors.
Complications
If phenylketonuria is not treated early and properly managed, high phenylalanine levels can cause serious complications.
Possible complications include:
-
Severe intellectual disability
-
Delayed development
-
Behavioral and emotional problems
-
Seizures
-
Tremors
-
Poor concentration
-
Neurological damage
Women with PKU who become pregnant without controlling their phenylalanine levels may also risk complications for the developing baby.
Prevention
Phenylketonuria cannot be prevented because it is a genetic condition. However, early detection and proper management can prevent serious health problems.
Preventive strategies include:
-
Newborn screening to detect PKU shortly after birth
-
Following a strict low-phenylalanine diet
-
Avoiding foods high in protein such as meat, eggs, and dairy
-
Avoiding products containing artificial sweeteners like aspartame
-
Regular blood tests to monitor phenylalanine levels
-
Genetic counseling for families with a history of PKU
With lifelong dietary management and medical monitoring, individuals with phenylketonuria can maintain good health and normal development.
Advertisement
